bringing data and tools together
Genboree is a web-based platform for multi-omic research and data analysis using the latest bioinformatics tools.
You can upload your data and perform various analyses using a “drag and drop” user interface. Keep it private or share with collaborators. Bioinformatics tools and computational infrastructure are available for researchers who may not have programming expertise, or the time to pursue technical programming and/or scripting.
Get Started
commons
Online collaboration
The Genboree Commons is a place to create projects, discussion forums and wikis, and to share documents with your colleagues.
All of your documents and content are private within a project unless you choose to share with others. It’s easy to add colleagues to your projects, change access privileges, and communicate with one another.
ClinGen
Clinical Genome Resource
One of the main goals of the Clinical Genome Resource project is to enable curators and researchers to understand the clinical significance of genes, variants and genetic disorders. We contribute to this effort by developing several software products, web services and user interfaces to facilitate curation of genes and variants.
Learn more about our software products developed for ClinGen

THE HUMAN
EPIGENOME ATLAS
A Data Repository of Tissue-specific Epigenomic States
The Human Epigenome Atlas contains human reference epigenomes and the results of integrative and comparative analyses. Atlas data provides detailed insights into locus-specific epigenomic states, including histone marks and DNA methylation across tissues, cell types, developmental stages, physiological conditions, genotypes, and disease states.
Research
 Image: National Institutes of Health
Image: National Institutes of Health
NIH ExRNA Communication Program
The goal of the ERCP is to better understand the fundamental biological mechanisms of extracellular RNA (exRNA) generation, secretion, and transport, to create a public dataset of where exRNAs exist in normal human body fluids, and to explore their potential as therapeutics and biomarkers.
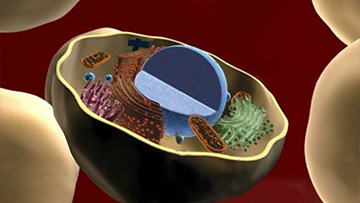 Image: National Institutes of Health
Image: National Institutes of Health
The NIH ClinGen Resource
One of the components of the Clinical Genome Resource project is ClinGenDB infrastructure to enable the development of a knowledge base about genetic variants of clinical significance. ClinGenDB infrastructure consists of databases and web services implemented using Genboree KnowledgeBase (GenboreeKB).
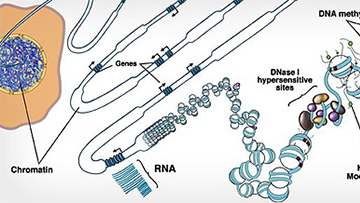 Image: National Institutes of Health
Image: National Institutes of Health
NIH Roadmap Epigenomics Mapping Consortium
The goal of this consortium is to map DNA methylation, histone modifications, chromatin accessibility, and small RNA transcripts in tissues and organs frequently involved in human disease.